



INTRODUCTION
According to the International Diabetes Federations‘ most recent estimate 382 million people (8.2% of the adults) suffer from diabetes mellitus (DM), and the number is expected to increase to 592 million people in less than 25 years. By widening the traditional glucocentric view subclinical inflammation and reactive oxygen and nitrogen species are more and more established as drivers of insulin resistance, beta cell dysfunction, and micro-/macrovascular complications. As brain insulin resistance is an early and common feature, Alzheimer disease (AD) has been hypothesised to be another form of diabetes. Diabetic encephalopathy is an increasingly explored complication of both, T1 and T2DM, which critically affects physical and mental integrity particularly in the elderly. Cerebral inflammation and oxidative stress triggered by recurrent hypoglycemia, C-peptide depletion (T1DM) and pro-diabetic risk factors (T2DM) are of key in mediating biochemical and structural brain changes affecting both neurons and astrocytes. Diabetes aggravates hepatic encephalopathy, and liver cirrhosis predisposes to diabetes. This overview will provide a comprehensive summary on the interaction between DM and liver disease in the evolution of cognitive and mental impairment. Special attention will be paid to molecular pathogeneses and its potential interference by current diabetes therapies.
DIABETES: A MECHANISTIC HUB FOR DISEASE CLUSTERING
Obesity and diabetes form a mechanistic hub for the evolution of chronic disease clusters and predispose to frailty and physical and mental disability. The connection between diabetes, cognitive impairment and dementia becomes increasingly recognised. Diabetes has been associated with a 44% acceleration of mental decline [1] and a 65% increase in the risk of incident Alzheimer disease (AD) [2]. Little is known about the interaction between diabetic and hepatic encephalopathy and how diabetes treatment regimens affect the patients' cognitive health.
DIABETIC ENCEPHALOPATHY (DE): SOME KEY FEATURES [3]
Cognitive dysfunction in DM was first recognised in 1922. In 1950 the term diabetic encephalopathy was introduced. Diagnostic criteria are not rigorously defined. They include
- Reduced performance of patients with T1DM or T2DM in one or more domains such as verbal memory, information processing speed, attention and executive functioning. Accompanied or not by subjective cognitive complaints.
- Cognitive deficits not directly related to acute dysglycaemic episodes and peripheral neuropathy. Absence of overt dementia.
Multiple interconnected pathogenetic drivers include
- Obesity (neuroinflammation), hypercholesterinaemia (altered APP/tau processing).
- Dysglycaemia, cerebral insulin/IGF-1 resistance (neurotrophic factor deprivation), hyperinsulinaemia (reducing cerebral A-beta clearence).
- Cytokines (TNFalpha, IL6), oxidative stress, glucocorticoids.
- Endothelial dysfunction, hypertension, atherosclerosis.
- C-peptide depletion (T1DM): inflammation, neuronal/oligodendroglial cell loss, gray / white matter atrophy.
COGNITIVE DECLINE: IMPACT OF AGEING AND T2DM [4]
Age-related cognitive decline in the general population:
- Slow cognitive decline from ~ 40 years old onwards.
- High variation among individuals.
Mild cognitive decline and incident dementia in T2DM:
- T2DM-related cognitive impairment: prevalent in all age groups.
- Cerebral correlates: brain atrophy, disturbed white matter integrity, vascular lesions → increased brain vulnerability to cerebro-vascular- and Alzheimer- type incidents in elderlies.
- Differentiated against the rapid cognitive decline preceding dementia.
- 50% increased dementia risk in T2DM → average earler onset of 2.5 years compared to people without DM.
VICIOUS CIRCLES IN HEPATIC ENCEPHALOPATHY [5]
HE in liver cirrhosis is a clinical manifestation of a low grade cerebral edema, related to an ammonia-induced exhaustion of the astroglial volume-regulatory capacity. A vicious circle between astrocyte swelling and ROS/RNOS triggers signals interfering with neuronal transmission. Impairments of synaptic plasticity finally lead to HE symptoms. Although HE has been considered as a reversible condition the recent demonstration of cerebral senescence may indicate a chronically progressive component possibly affecting the brain reserve as does DE.
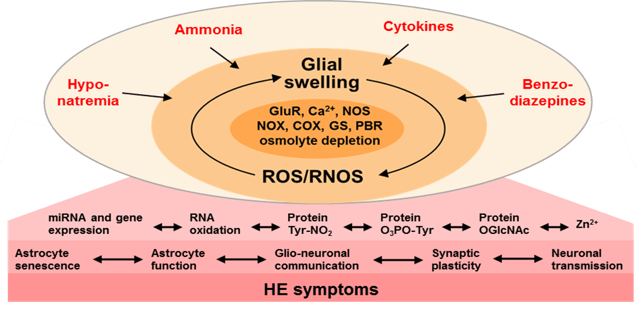
Figure: Vicious circles in hepatic encephalopathy (HE). Modified from Häussinger, D. & Schliess, F. 2008, Gut 57:1156-1167. GluR: glutamate receptor, NOS: nitric oxide synthase, COX: cyclooxygenase, GS: glutamine synthetase, PBR: peripheral type benzodiazepine receptor, ROS/RNOS: reactive oxygen and nitrogen species.
DE & HE: SIMILARITIES, SYNERGISMS, AND DIFFERENCES [6]
Similarities
- Diversity of triggering factors and conditions.
- Partly overlapping pathogenic mediators (altered BBB permeability, micro- and astroglia, ROS/RNOS, neuroinflammation, altered synaptic plasticity and functional connectivity between brain areas).
Synergisms
- Multiple interactions between underlying disease entities such as fatty liver, NASH, liver cirrhosis, and (hepatogenous) diabetes.
- Hepatogenous diabetes: secondary to liver cirrhosis, peripheral insulin resistance, hyperinsulinaemia related to portocaval shunting and decreased insulin degrading enzyme activity in the liver.
- Cirrhotic diabetics: prone to more severe recurrent HE episodes due to
- Augmentation of (subclinical) inflammation.
- Synergistic action of HE precipitants (ammonia) with the triggers of DE
- Increased ammonia exposure by a diabetes-related increase of glutaminase activity (liver, small intestine, kidney), a delayed gastrointestinal transit and intestinal overgrowth & translocation of bacteria.
- Diabetic cirrhotics with hepatitis C virus (HCV) infection: more severe HE at earlier stages of biochemical decompensation. Higher HE risk even in the absence of visceral bleeding and infection. Direct crosstalk between HCV and insulin signaling.
Differences
- Course: mild but progressive (DE) vs. highly variable and dynamic (HE).
- Treatment: not specifically addressed within established CVD risk and DM management (DE). Acute amelioration by switching of triggers (HE).
POTENTIAL IMPACT OF ANTI-HYPERGLYCAEMIC TREATMENTS ON DE/HE
Metformin [7]
- Prevented HE in diabetic cirrhotic patients, explained by partial inhibition of glutaminase, amelioration of inflammation and improved insulin sensitivity.
- Reduced thrombospondin-1 suppression by ammonia in rat astrocytes → impact on synaptic plasticity in HE?
GLP-1 receptor agonists [8]
- Preclinical evidence for restoration of cerebral insulin sensitivity, decreasing microglial activation and A-beta in insulin resistant neurons, provision of neuro-protection, promotion of neurogenesis, restoration of synaptic plasticity and neuropsychological improvement in people with mild cognitive impairment and neurodegenerative disorders.
Intranasal insulin [9]
- Improves neuropsychological performance in people with mild cognitive impairment and early dementia, possibly by compensating for cerebral insulin resistance, thereby restoring regulation of neuronal plasticity by insulin.
11-beta hydroxysteroid dehydrogenase (HSD)1 inhibitors [10]
- Reduce the cortisol load associated with T2DM and ageing. Improve neuropsychological performances in T2DM and ageing animal models and also in elderly people and people with T2DM, which is consistent with 11-beta HSD1 expression in human brain.
CONCLUSION
DE and HE relate to different but mutually interacting disease entities. They share common cerebral mediators. DM provokes HE in liver cirrhosis, which again triggers DM. Neuropsychological impairment in DM as well the interference of anti-hyperglycaemic compounds with mild cognitive impairment and dementing processes are increasingly gaining attention. However, defining the impact of established and novel diabetes treatments specifically on DE and HE requires a more targeted clinical exploration.
REFERENCES
[1] Arvanitakis, Z. et al. 2004, Arch. Neurol. 61:661-666. [2] Talbot, K. et al. 2012, J. Clin. Invest. 122:1316-1338; Duarte, A.I. et al. 2013, Biochem. Biophys. Acta 1832:527-541. [3] Biessels, G.J. et al. 2014, Lancet Diabetes Endocrinol. 2:246-245; Meng, X.F. et al. 2014, Mol. Neurobiol. 49:673-684; Cai, D. 2013, Trends Endocrinol. Metab. 24:40-47; Talbot, K. et al. 2012, J. Clin. Invest. 122:1316 -1338; Wahren, J. et al. 2012, Diabetes 61:761-774; Sima, A.A.F. 2010, Acta Diabetol. 47:279-293; Minhout, G.S. et al. 2006, Diabetologia 49:1447-1448. [4] Biessels, G.J. et al. 2014, Lancet Diabetes Endocrinol. 2:246-245. [5] Görg, B. et al. 2014, GLIA, 2013, Arch. Biochem. Biophys. 536:158-163; Schliess, F. et al. 2006, Biol. Chem. 1363-1370; Häussinger, D. et al. 1994, Gastroenterology 107:1475-1480; Norenberg, M.D. et al. 1992, Prog. Brain. Res. 94:261-269. [6] Ahmadieh,H. & Azar, S.T. 2014, Diabetes Res. Clin. Pract. 104:53-62; Ampuero, J. et al. 2013; Metab. Brain Dis. 28:277-279; Butterworth, R.F. 2013, Nat. Rev. Gastroente-rol. Hepatol. 10:522-528; Gundling, F. et al. 2013, Digestion 87:75-84; Purkayastha, S. & Cai, D. 2013, Mol. Metab. 2:356-363; Lonardo, A. et al. 2009, Expert Rev. Anti Infect. Ther. 7:293-308; Häussinger, D. & Schliess, F. 2008, Gut 57:1156-1165; Sigal, S.H. et al. 2006, Am. J. Gastroenterol. 101:1490-1496. [7] Jayakumar, A.R. et al. 2014, J. Neurochem.; Ampuero J. et al. 2012, PLoS ONE 7:e49279; Browning, J.D. & Horton, J.D. 2004, J. Clin. Invest. 114:147-152. [8] Talbot, K. & Wang, H.-Y. 2014, Alzheimer‘s Dement. 10:S12-S25; Corbett, A. et al. 2012, Nat. Rev. Drug Discov. 11:833-846. [9] Freiherr, J. et al. 2013, CNS Drugs 27:505-514; Shemesh, E. et al. 2012, J. Clin. Endocrinol. Metab. 97:366-376; Heinemann, L. 2011, Int. J. Clin. Pract. 170:31-46; Arnolds, S. & Heise, T. 2007, Best Pract. Res. Clin. Endocrinol. Metab. 21:555-571. [10] Heise, T. et al. 2014, Diabetes Obes. Metab. 16:1070-1077; Strachan, M.W. et al. 2009, Diabetes Obes. Metab. 11:407-414; Sandeep, T.C. et al. 2004, Proc. Natl. Acad. Sci. 101:6734-6739.