



As previously reported, the Clinical Trial Information System (CTIS) can help to get a faster approval for a multi-national, multicentre Phase III Study (read here part 1 and part 2 of the REGULATION (EU) No 536/2014 blog series).
However, for Profil as a CRO performing predominantly early phase studies, the timelines foreseen within CTIS are not such an advantage. Therefore, our department of Regulatory Affairs started very early to participate in trainings, information sessions and discussions about CTR and CTIS. Furthermore, to strengthen the conduct of early phase studies in Germany, the German authorities and the ethics committees have been asked to review the approval timelines for monocentric mono-national clinical studies. Although the timelines cannot be adapted within CTIS, there might be recommendations to adhere to as published recently by BfArM [1].
After having submitted either part 1 and 2 ourselves or being the supporting team for a submission performed by a Sponsor or another CRO, we noticed the following general difficulties:
- Notices and alerts are only provided via CTIS (not noticeable which notices and/ or alerts have been processed already)
- Timetable is not adapted if responses or requests are entered more rapidly than requested
- Handling of redacted documents quite tedious (first “for publication” then the redacted version), especially if documents need to be exchanged again
- If a Request for Information (RFI) is raised for a part 1 document only (e.g. the protocol), but might have an impact on a part 2 document (e.g. the ICF), this document (the ICF) cannot be adapted in one step (due to a missing functionality in CTIS), but requires a substantial modification
- Complicated download of many (partly unnecessary) documents of a Request for Information (RFI) or report
For a submission in CTIS, two approaches might be used: the organisation-centric approach or the trial-specific approach.
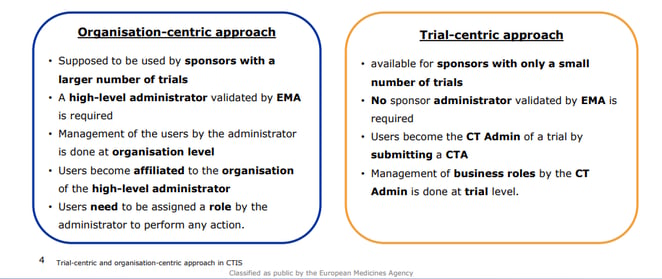
Organisation- and trial-centric approach in CTIS [2]
At Profil, we usually work with industrial sponsors. Hence, up to now we used the organisation-centric approach. However, this approach seems to be applied automatically by the system based on the existing data stored in CTIS [2].
The timelines from submission to approval for our early phase studies might take nearly 3 months in case of complex RFIs. Nevertheless, the requests we received so far were easy to implement and more of administrative nature than a contextual challenge. The advantage of the organisation-centric approach is the potential “work-sharing”. As stated above, it is possible to take over the whole responsibility for the submission on behalf of a Sponsor or the different tasks can be allocated to different stakeholders, either at the Sponsor’s organisation or at the supporting CRO.
Looking at the RFIs we received so far, we recommend considering the following:
- An often referenced document in the clinical trial protocol (not yet submitted) might be requested
- Blood volume should be indicated in tea- or tablespoons
- Fields in CTIS not marked with an asterisk as “mandatory” might be requested to be filled out, though
- The GDPR statement as provided by EMA might be considered not sufficient to cover German expectations
- The quantification of possible side effects should be clear – e.g. what is meant with "in rare cases"
Needless to say that all stakeholders are still gathering experience, are adapting internal processes and SOPs, and putting together all available information to have as many advantages as possible from a submission in Europe, including Germany.
Read more about our expertise in testing medical technology: Insulin pump development and design, artificial pancreas study and the AP@home project, glucose monitoring.