



The role of insulin and c-peptide in physiological molecular pathways
Beta cell integrity plays an essential role in blood glucose homeostasis and the development of diabetes mellitus. The formation of insulin in the beta cell is preceded by the biosynthesis of the single chain pro-hormone proinsulin. Proinsulin is stored in vesicles within the beta cell and contains the insulin A and B chain agglutinated by a connecting peptide (C-peptide). In case of insulin requirement, the proinsulin within the vesicles is cleaved into insulin and C-peptide by the catalytic activity of the carboxypeptidase H and the pro-protein convertases 1 and 2. Beyond metabolic effects, insulin and C-peptide trigger important signalling pathways involved in physiological regulation of endothelial function and vascular biology.
As shown in figure 1, in metabolic target cells, like adipocytes or muscle cells, the activation of the insulin receptor (IR) initiates a complex downstream signalling cascade through the activation of phosphatidylinositol-3-kinase (PI3K) releasing the downstream of glucose molecules into the cell by the translocation of the glucose transporter GLUT 4 into the cell membrane.
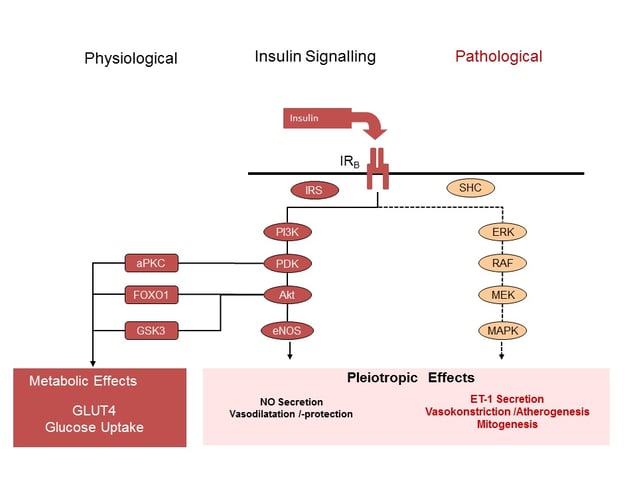
Figure 1: Schematic presentation of intracellular insulin signalling in difference tissues. Presentation of PI3K and MAPK signalling cascades following binding of insulin to the insulin receptor subtype B. (aPKC = atypical Protein-Kinase C; eNOS = endothelial Nitric Oxide Synthase; ET1 = Endothelin 1; FOXO1 = Forkhead Box01; IR = Insulin Receptor; IRS = Insulin Receptor Substrate; MAPK = Mitogen Activated Protein-Kinase; MEK = dual threonine and tyrosine recognition kinase; NO = Nitric Oxide; PDK = Phosphoinositide-Dependent Kinase; PI3K = Phosphatidylinostol 3 Kinase; ; RAF =Proto-Oncogenes; SHC = Adapter Protein).
In contrast, in non-metabolic cells, like endothelial cells, the activation of the same PI3K signalling pathway leads to an increase in nitric oxide (NO) release and subsequent vascular smooth muscle relaxation. Moreover, NO interacts with the expression of adhesion molecules (VCAM-1, ICAM-1, E-Selectin), proinflammatory activity, and platelet aggregation (1,2,3).
Paradoxical roles of insulin and c-peptide in pathological pathways
Under pathological conditions, like insulin resistance, the phosphorylation of the insulin receptor will cause vasoconstriction by the activation of an alternate signalling pathway within endothelial cell and the release of endothelin 1 (ET-1) instead of NO. The activation of this alternate insulin signalling pathway (MAPK/ERK pathway) is made responsible for the paradox observations with insulin causing vasoconstriction and mitogenic signalling. Therefore, the biological effects of insulin differ depending on the target cell and the biological signalling pathways activated under certain conditions (4,5,6,7).
Obesity and the activity of visceral adipocytes are major gate keepers in the augmentation of insulin resistance and pro-atherogenic pathways. Paradoxical effects of insulin in microvascular blood flow have been observed in obese subjects (8), patients with type 1 diabetes (9,10), patients with type 2 diabetes (11,12,13,14) and female patients with polycystic ovarian syndrome (15). As shown in figure 2, mediators released from the visceral adipocytes like angiotensin II, free fatty acids, or TNF-alpha shift the insulin signal within the endothelial cell. Insulin action through PI3K signalling is blunted while its signals through the MAPK/ERK pathway remain intact or even enhanced, tilting the balance between ET-1 and NO. Microvascular insulin resistance and dysfunction not only decrease tissue perfusion and increase the risk of end organ damage, it also contribute to the development of hypertension. There is increasing evidence that also the pharmacokinetic properties of external insulin formulation might have distinct effects on insulin signalling pathways with varying effects on microvascular function and blood flow (16, 18,19).
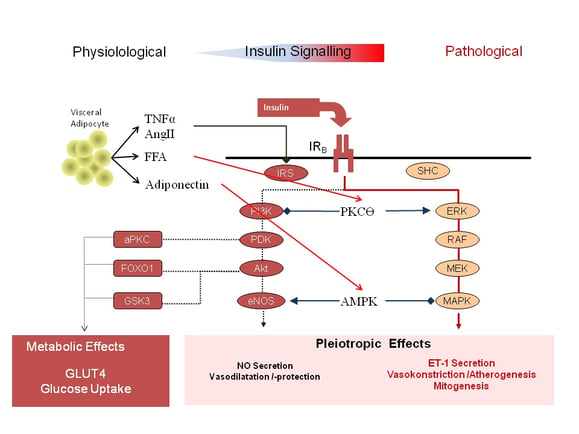
Figure 2: Shift in intracellular insulin signalling from the PI3K pathway to the MAPK/ERK pathway by mediators from visceral adipocytes (AngII= Angiotensin II; aPKC = atypical Protein-Kinase C; eNOS = endothelial Nitric Oxide Synthase; ET1 = Endothelin 1; FFA = Free Fatty Acids; FOX01 = Forkhead Box01; IR = Insulin Receptor; IRS = Insulin Receptor Substrate; MAPK = Mitogen Activated Protein-Kinase; MEK = dual threonine and tyrosine recognition kinase; Mitogen Activated Protein-Kinase; NO = Nitric Oxide; PDK = Phosphoinositide-Dependent Kinase; PI3K = Phosphatidylinostol 3 Kinase; RAF =Proto-Oncogenes; SHC = Adapter Protein; TNFα = Tumor Necrosis Factor α).
As illustrated in figure 3, progression of type 2 diabetes mellitus is driven by deteriorating beta cell function and an increase in the release of proinsulin. Numerous studies suggested an association between increased proinsulin levels and the development of vascular complications in subjects with or without diabetes (20,21, 22,23,24,25). Even though the exact mechanism how proinsulin might exaggerate vascular pathology is not fully solved, there is increasing evidence that proinsulin stimulates the MAPK/ERK signalling pathway in endothelia cells.
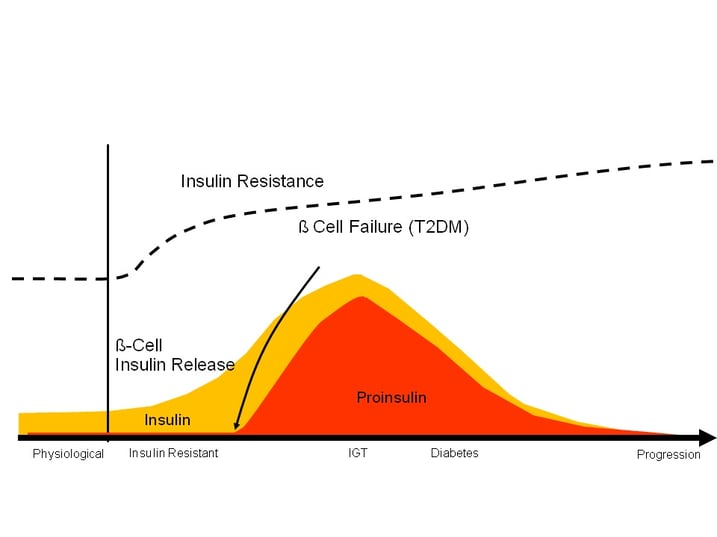
Figure 3: Schematic presentation of the time course of insulin resistance, insulin and pro-insulin secretion from insulin resistance to pathological glucose tolerance and diabetes mellitus type 2.
As shown in figure 4, proinsulin was found to bind with high affinity to an isoform of the insulin-receptor (IRA) incorporated into the cell membrane of numerous different cell types like endothelial cells. Activation of this insulin receptor subtype predominantly activates the MAPK/ERK downstream signalling cascade (26), thereby driving the vasoconstrictive, pro-artherogenic, and mitogenic properties of proinsulin. In addition, pro-insulin raises plasminogen activator inhibitor-1 (PAI-1) levels (27,28,29), thereby accelerating the high pro-thrombotic potency observed in patients with T2DM.
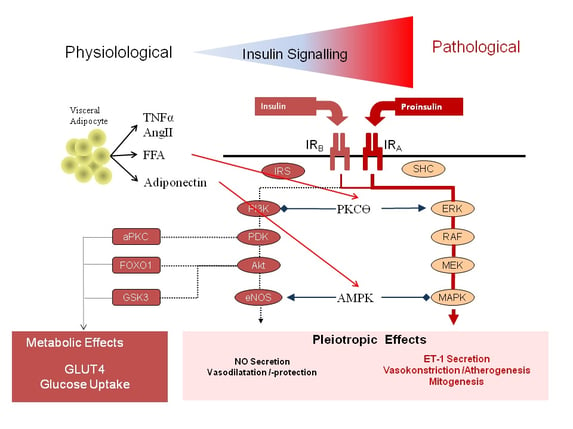
Figure 4: Shift in intracellular insulin signalling from the PI3K pathway to the MAPK/ERK pathway through activation of the insulin receptor subtype A (AngII= Angiotensin II; aPKC = atypical Protein-Kinase C; eNOS = endothelial Nitric Oxide Synthase; ET1 = Endothelin 1; FFA = Free Fatty Acids; FOX01 = Forkhead Box01; IR = Insulin Receptor; IRS = Insulin Receptor Substrate; MAPK = Mitogen Activated Protein-Kinase; MEK = dual threonine and tyrosine recognition kinase; NO = Nitric Oxide; PDK = Phosphoinositide-Dependent Kinase; PI3K = Phosphatidylinostol 3 Kinase; RAF =Proto-Oncogenes; SHC = Adapter Protein; TNFα = Tumor Necrosis Factor α).
In a couple of studies it was shown that treatment of type 2 diabetes mellitus with incretin based interventions (GLP-1 receptor agonist or DPP-IV inhibitors) improve beta cell function and diminishes the release of pro-insulin from the beta cells, while treatment with sulfonylureas evolves the opposite effect (30,31,32) Therefore, despite comparable blood glucose control, distinct pharmacological interventions might exert fundamental different effects on beta cell function and the entailing vascular pathology in patients with type 2 diabetes mellitus.
C-peptide is an important split product accruing through the conversion process from pro-insulin to insulin. While over decades C-peptide was believed to be a split product without any biological activity recent investigations identified C-peptide as regulatory peptide within the microcirculation (33,34,35,36). As shown in figure 5, C-peptide affects microvascular blood flow by interfering with several signaling pathways in the endothelial cell. Recent data suggest, that C-peptide binds to a G-protein coupled receptor, increasing Ca2+ influx with subsequent activation of Ca(2+)-calmodulin. C-peptide supplementation in subjects with type 1 diabetes was shown to stimulate eNOS and the Na+/K+-ATPase in erythrocytes and endothelial cells.
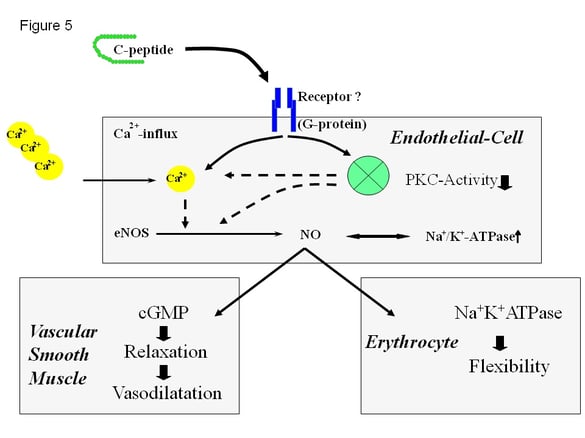
Figure 5: Recent understanding of the molecular effects of C-peptide on endothelial cells, vascular smooth muscle cells, and blood rheology. It is postulated that C-peptide binds to a specific receptor in the cell membrane with subsequent intracellular activation of a G-protein coupled signalling pathway (Ca2+ = Calcium ions; cGMP = cyclic guanosin mono phosphate; eNOS = endothelial nitric oxide synthase; Na-KATPase = sodium potassium ATPase; = nitric oxide; PKC = protein kinase C; activity)
Recent clinical studies in subjects with type 1 diabetes suggested beneficial effects of C-peptide supplementation on microvascular complications like nephropathy or neuropathy (37,38). Nevertheless, the effects of C-peptide in type 2 diabetes mellitus and the development of atherogenesis still appear contradictory. Progression of insulin resistance is temporarily linked to an increased release of insulin and C-peptide from the beta cell. In patients with diabetes mellitus type 2, an increased deposition of C-peptide was found in early arteriosclerotic lesions located directly adjacent to monocytes and CD4 positive lymphocytes (39,40). Therefore it seems conceivable, that C-peptide in excessive concentrations, like in patients with type 2 diabetes, might evolve pro-atherogenic effects, while the lack of C-peptide, like in patients with type 1 diabetes, might contribute to the development of microvascular complications.
What does all that mean?
In conclusion, diabetes mellitus is associated with several vascular microvascular complications like retinopathy, nephropathy, and neuropathy. The role of the beta cell in the development of type 1 and type 2 diabetes mellitus is well established. Beside its pathophysiological role in the derangement of blood glucose control, there is increasing evidence that disturbances in beta cell function might play a direct causal role in the development of microvascular complications associated with diabetes mellitus. Further research is required to understand the effect of different pharmacological interventions on beta cell physiology and the development of associated microvascular complications.