



Waiving Bridging studies under certain circumstances for biosimilar applications?
A bridging study is a study performed in a new region to provide pharmacodynamic or clinical data on efficacy, safety, dosage and dose regimen that will allow extrapolation of foreign clinical data to the population in the new region [1]. However, in most cases bridging studies between an original product versus a so called foreign reference or a local reference product generate costs without providing any notable benefit for the specific patient, nor notable scientific output.
The International Generic and Biosimilar Medicines Association (IGBA) [2] proposed in 2018 that bridging studies for biosimilar medicines application could be waived under certain circumstances. The proposal is based on an article by Webster and Woollett [3] published in 2017 dealing with the fact that authorities in general are open to accept data coming from publicly available information for the above mentioned reference products. Moreover, these studies are not legally required, but are requested at the discretion of the respective local authority (s. Figure 1).
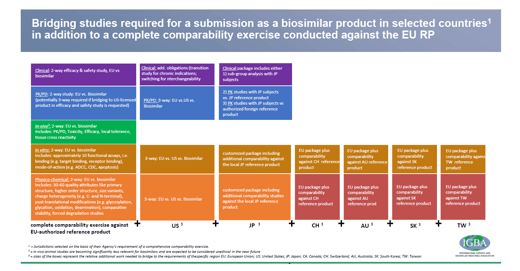
Figure 1: Bridging studies under different regulatory areas (click to get an enlarged view)
The existing international agreements between authorities such as the US FDA, the European Commission DG Santé and the EMA could serve as templates for confidentiality commitments between other regulatory agencies. Based on these agreements, data related to approved drugs can be exchanged and taken into account for the review of a new biosimilar product.
However, to communicate in a clear and transparent way some definitions are needed, e.g.:
Definition of a Reference Product: any originator product that has been approved by a Stringent Regulatory Authority (SRA/ WHO-Listed Authority (WLA) (Maturity Level 4( ML4) [4], based on a stand-alone dossier.
Definition of a Comparator Product: a reference product used for head-to-head similarity /comparability study to support the biosimilar application and approval.
Definition of a Global Comparator product: any originator biological product authorized by any SRA/ WHO-Listed Authority (WLA ML 4) and qualified as Comparator Product by complying with pre-specified criteria. It can be used for biosimilar product development to support a biosimilar application and approval in any jurisdiction around the world.
In case a local biosimilar application occurs, the Reference Product is a so-called Local Reference, this can also apply to a Comparator Product if a locally –approved product is used for the comparison. Another option would be to use a non-locally-approved Reference product, then called a Foreign Reference.
To qualify as a Global comparator the kind of authority approving the product is important as well as the manufacturing steps and sites (a comparator product facility must have been inspected for compliance with cGMPs, cGCPs, CGLPs and cGDPs). Furthermore, it has to be fully identifiable and an evaluation report should be ideally publicly available. One option to get to know which comparator product might be used, could be that WHO publishes in cooperation with SRAs a list of potential global comparator products. This option is supported by the increasing collaboration amongst regulators as EMA, FDA, HC and PMDA as well as by establishing clusters and fora to share regulatory practices and information on medicinal products, e.g. International Pharmaceutical Regulators Programme/IPRP [5], WHO-International Conference of Drug Regulatory Authorities/ICDRA, WHO Pilot Prequalification Procedure for Biotherapeutic Products: trastuzumab and rituximab).
Another valuable tool is the ISO Identification of Medicinal Products (IDMP) standard [6]. Within this worldwide system for establishing unique identifiers for medicinal products a reliable exchange of information on medicinal products is feasible. Four domains of master data will be implemented in a phased programme: substance, product, organization and referential (SPOR) data.
Waiving bridging studies between the locally approved Reference product and the non-locally-approved Reference product (Foreign Reference) could be considered in case the Foreign Reference meets the following requirements:
- is defined as a Comparator Product
- contains the same active pharmaceutical ingredient (API) and has the same pharmaceutical form and same route of administration
- has the same composition of excipients
- was approved in the respective jurisdiction based on essentially the same original data package as the locally-approved reference product
- subsequent manufacturing changes were regulated according to ICH Q5E principles [7].
Whether a bridging can be waived has certainly to be evaluated on a case by case basis together with the responsible regulatory authority. However, it is important to know that regulatory authorities tend to follow a certain reasoning when considering to waive a certain clinical trial.