



Reevaluation of glucose-dependent insulinotropic polypeptide (GIP) pharmacology in combination with glucagon-like peptide-1 (GLP-1) for metabolic diseases: Overview on biological GIP and GLP-1 effects at the organ/tissue level.

Fig. 1
Dual agonism of GIP and GLP-1 receptors is a promising new approach currently in clinical development for the treatment of diabetes, obesity, and associated complications [1]
The incretin effect defines the main physiological role of GIP and GLP-1
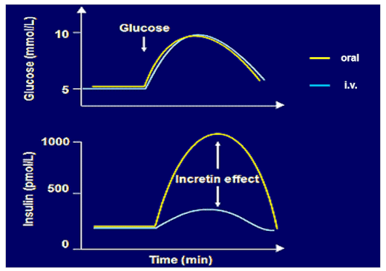
GIP and GLP-1 increase insulin secretion following nutrient-induced secretion from the gut (incretin effect): There is a stronger insulin secretion response to oral compared to 'isoglycemic' intravenous glucose administration due to secretion and action of incretin hormones.
Both GIP from intestine K- and GLP-1- from intestine L-cells are mainly stimulated by the ingestion and absorption of carbohydrates and triglycerides or their digestion products. A functioning enteroinsular axis is essential for the maintenance of normal glucose tolerance.
Enteroinsulin axis
One of the main differences between GIP and GLP-1 is their influence on glucagon secretion: GLP-1 suppresses glucagon secretion, and GIP increases it, both in a glucose-dependent manner. Whereas GLP-1 can even stimulate glucagon at PG levels ≤ 90 mg/dL. [2] GIP has the potential to increase glucagon secretion without hypoglycemia. While the classical incretin concept is based on the assumption that incretin hormones such as GIP and GLP-1 reach their main endocrine target cells via the bloodstream, there is some evidence that GIP (and possibly GLP-1) can be produced in relatively small amounts in endocrine alpha-cells of the pancreas [3]. From there, they can exert paracrine effects on neighbouring beta-cells and other endocrine cells. GIP receptors are found on alpha-cells and also on delta-cells and polypeptide gamma-cells of the endocrine pancreas. GIP and GLP-1 receptors are also abundantly expressed on beta-cells in the islets of Langerhans.
GIP-induced insulin secretion and consequences for the incretin effect
Endogenous GIP affects postprandial plasma glucose excursions and insulin secretion more than endogenous GLP-1, but the hormones contribute additively to postprandial glucose regulation in healthy individuals [4]. However, in people with type 2 diabetes, GIP does not contribute at all to the total insulin secretion effect of the combination while the insulinotropic effect of GLP-1 is only slightly impaired. This may also be referred as ‘GIP resistance’: a significantly reduced ability of GIP to stimulate insulin secretion even in hyperglycemia.
There are competing hypotheses to explain the GIP resistance. One explanation assumes a specific reduction in GIP receptor expression or a disrupted GIP post receptor or impaired GIP post receptor signal transduction in people with diabetes mellitus [5]. Furthermore, Ectopic lipid deposition or inflammation in the endocrine pancreas, could potentially influence GIP receptor expression.
Body weight regulation in the brain
GLP-1 at pharmacological concentrations reduces appetite, food intake and - in the long term - body weight, and a similar role is developing for GIP, at least in animal studies. There is no obvious change in resting energy expenditure in response to GIP stimulation.
Receptors for GIP and GLP-1 have been identified in brain regions involved in the regulation of appetite, satiety, food/energy intake and energy expenditure [6]
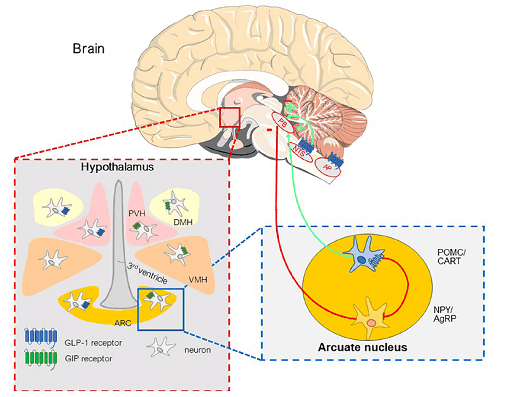
Fig. 1: GIP and GLP-1 receptors in the hypothalamus and brainstem involved in the regulation of energy intake (meal initiation and termination) and body weight. PB: Parabrachial nucleus; NTS: Nucleus tractus solitarii; AP: Area postrema; PVH: Para-ventricular hypothalamus; DMH: Dorso-medial hypothalamus; VMH: Ventro-medial hypothalamus; ARC: Arcuate nucleus; POMC/CART: Proopiomelanocortin/cocaineamphetamine-regulated transcript;NPY: Neuropeptide Y; AgRP: Agoutirelated peptide.
Significant progress has been made in identifying nuclei in the hypothalamus and brainstem and their role in initiating and terminating meals that determine energy balance and body weight regulation [7].
GIP and/or GLP-1 receptors are possibly involved in anti-apoptotic effects, synaptic plasticity, memory, reward functions, and emotional responses, which may have a positive impact on a number of neurodegenerative diseases [8].
Adipose tissue
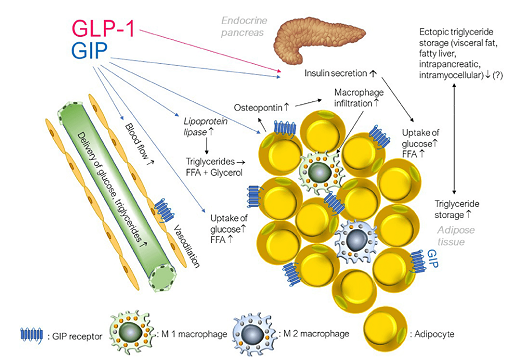
GIP, but not GLP-1, increases triglyceride storage in white adipose tissue not only by stimulating insulin secretion but also by interacting with regional blood vessels. GIP has been shown to increase glucose uptake, lipoprotein lipase activity and lipogenesis (i.e. re-esterification of free fatty acids to triglyceride) [9]. GIP also supported de novo lipogenesis from glucose as well as lipolysis (the mobilisation of FFA in the fasting state).
Some groups interpret the lipotropic function of GIP as positive and argue that it can support the expansion of healthy subcutaneous fat deposits [10] while most studies consider GIP to be an adipogenic endogenous hormone in adipose tissue [11]. Data suggest that GIP worsens insulin sensitivity through effects on cytokine production [12] in adipose tissue [13]. However, limited GIP-induced acute postprandial release of cytokines may represent a physiological response required for healthy adipose tissue function [14]. It can only become pathogenic when exaggerated, as in insulin-resistant obesity [15].
The expression of GIP receptors in vascular adipose tissue in endothelial cells and monocytes/macrophages probably influence each other and increase inflammation [16]. However, significant global overexpression of GIP receptors reduced inflammatory markers in mice and prevented obesity [17].
Patterns of effects of GIP receptor agonism or antagonism
There is currently an unresolved paradox of similar effects of GIP agonists and antagonists on metabolism. Both GIP receptor agonism and antagonism were able to reduce body weight or prevent diet-induced obesity in mice [18].
A recent review by Campbell [19] attempts to reconcile the paradoxical observations. There might be a compensatory relationship between incretin receptors and how GIP-receptor antagonism can enhance GLP-1 receptor activity. Furthermore, chronic GIP-receptor agonism can lead to desensitization and eventual loss of GIP receptor activity, which then mimics antagonism. Overall, it is clear that a deeper understanding of GIP biology is required to understand how modulating this system impacts metabolic homeostasis.
Improvement in insulin sensitivity
The recent finding that GIP/GLP-1 receptor co-agonists like tirzepatide have superior efficacy compared to selective GLP-1 receptor agonists with respect to glycemic control as well as body weight has renewed interest in GIP [20], which previously was thought to be without any therapeutic potential. The assessment of the homeostasis model also suggests an improvement in insulin sensitivity in type 2 diabetics treated with tirzepatide that is significantly greater than that seen with treatment with a selective GLP-1 receptor agonist. This can only be partially attributed to weight loss [21] reported a weight-independent effect of GIP receptor stimulation on insulin sensitivity. A recent study by Samms et al. [22] reported a weight-independent effect of GIP receptor stimulation on insulin sensitivity. From a mechanistic point of view, glucose uptake into brown adipose tissue seems to play an important role.
Further incretin-based drug development
For both GIP and GLP-1, positive effects were also observed on cardiovascular complications [23], indicating a therapeutic potential beyond the improvement of diabetes complications.
One focus of the recent research is into the long-term interaction of GIP and GLP-1 receptor signalling, which could potentially be clarified if a long-acting GIP receptor agonist, which has already been investigated in rodent studies [24,25], can also be studied in humans. Whether such a GIP receptor agonist [26, 27, 28, 29] has its own therapeutic potential currently remains an open question. Together with the already available GIP receptor antagonists it may be possible to clarify further questions that have hitherto remained unanswered. A detailed knowledge of incretin physiology and pathophysiology will be a prerequisite for designing more effective incretin-based drugs for metabolic diseases.