



Introduction
The actual Medical Device Directive (MDD) 93/42/EEC [1]and the Active Implantable Medical Device Directive (AIMDD) 90/385/ECC [2]are the basic directives for all kinds of medical devices in Europe. These directives are mandatory for all member states and have to be put into national legislation by the national parliaments within a given time limit. It is not allowed to reduce or change the requirements of the directive but the parliaments can implement additional requirements like the German “Medizinprodukteberater” (consultant for medical devices) in §31 of the German medical device law.
In contrast to the Medical Device Directive the Medical Device Regulation (MDR) [3]comes directly from the European Commission in Brussels without any approval by the national parliaments and has to be applied as European, supranational law within a given time limit. Additional national requirements resolved by the national parliaments are possible.
One of the reasons for the new MDR was the PIP-scandal: One manufacturer used the cheaper industrial silicon for breast implants instead of the ultra-pure medical silicon. Once this scandal had been made known publicly the Commission decided to tighten up the directive to prevent this kind of criminal process.
Changes
The actual MDD 93/42/EEC has 60 pages including 23 articles (24 pages) and 12 Annexes (36 pages). The new MDR consists of 333 pages with 123 articles (92 pages) and 17 annexes (241 pages). The main changes or new content of the MDR are:
- The manufacturer hat to appoint a qualified person in the company which has qualified know-how in the field of medical devices.
- The European EUDAMED database will be broadly enlarged.
- The activities and the review certification of the notified bodies will be harmonized Europe-wide (MDR certificate)
- There will be a change in the conformity assessment procedure. A procedure comparable with Annex VI of the MDD is no longer available.
- A so-called Scrutiny Procedure was implemented: The notified bodies can be committed to report any new application for a conformity assessment for a high-risk product to the medical device coordination group (MDCG) which decides on the clinical evaluation of the product.
- The classification of some products will change: Some of the implantable products which are now class IIb products (MDD) will meet the requirements for class III products and software will rarely be a class I product.
- The requirements for reprocessing of disposable products have increased.
- A more detailed specification of the technical documentation of the medical device was included in Annex II of the MDR. A new request of the MDR is the continuous update of the documentation.
- The retention period of the technical documentation was doubled from 5 years for the MDD to 10 years for the MDR.
- Each medical product needs a unique device identification (UDI). With this identification system the Commission plans an obligation for identification and registration of medical products.
- There are new requirements for labelling of medical devices.
- Clinical evaluation and clinical investigation will be regulated and prescribed in more detail.
- The clinical evaluation has to be updated with post market information of the now important post market surveillance.
- The following figure shows the dependencies between documents under the MDR:
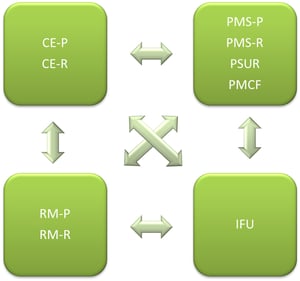
CE-P: Clinical Evaluation Plan
CE-R: Clinical Evaluation Report
PMS-P: Post Market Surveillance Plan
PMS-R: Post Market Surveillance Report
PSUR: Periodic Safety Update Report
PMCF: Post Market Clinical Follow Up
IFU: Instructions for Use
RM-P: Risk Management Plan
RM-R: Risk Management Report
Consequences
The intention of the new MDR was to tighten up the MDD to make the medical products safer and more reliable. By upgrading the existing directives to a European regulation the conformity assessment procedure will be a uniform process in all member states which makes the results consistent and more transparent. To achieve that the MDR will put more emphasize on documentation for the manufacturer of a medical product. This will lead to rising costs for the final MDR-conform medical product. Additional cost will come due to the obligation for a new conformity assessment procedure for medical products already approved under the MDD, additional effort for post market surveillance and post market clinical follow-up as well as for the Compliance Officer. For small and medium enterprises there will be the risk that they do not have the necessary man power and/or the necessary financial resources to be compliant with the new regulation. As a consequence there is the risk that many small and medium enterprises and their products (!) will disappear from the market.